在Matlab中,我想
绘制一个氨基酸序列
图的
seqlogo图 。但是,我不希望通过熵来缩放绘图列的高度,而是希望所有列都具有相同的高度。
我正在根据
该问题的答案来修改代码,但我想知道seqlogo的参数或其他我遗漏的其他函数是否可以使列高保持一致。
另外,是否可以对序列配置文件进行统计转换以破解所需的输出? (列高均匀,每个字母的高度与其在seqprofile中的概率成线性比例)
回答:
解决此问题的最简单方法可能是直接修改
生物信息学工具箱功能SEQLOGO的代码(如果可能)。在R2010b中,您可以执行以下操作:
edit seqlogo 该功能的代码将显示在编辑器中。接下来,找到以下几行(第267-284行)并将其注释掉或将其完全删除:
S_before = log2(nSymbols); freqM(freqM == 0) = 1; % log2(1) = 0 % The uncertainty after the input at each position S_after = -sum(log2(freqM).*freqM, 1); if corrError % The number of sequences correction factor e_corr = (nSymbols -1)/(2* log(2) * numSeq); R = S_before - (S_after + e_corr); else R = S_before - S_after; end nPos = (endPos - startPos) + 1; for i =1:nPos wtM(:, i) = wtM(:, i) * R(i); end 然后将此行放在他们的位置:
wtM = bsxfun(@times,wtM,log2(nSymbols)./sum(wtM)); 您可能希望将文件保存为新名称,例如seqlogo_norm.m ,因此您仍然可以使用原始的未修改
SEQLOGO函数。现在,您可以创建序列轮廓图,将所有列标准化为相同的高度。例如:
S = {'LSGGQRQRVAIARALAL',... %# Sample amino acid sequence 'LSGGEKQRVAIARALMN',... 'LSGGQIQRVLLARALAA',... 'LSGGERRRLEIACVLAL',... 'FSGGEKKKNELWQMLAL',... 'LSGGERRRLEIACVLAL'}; seqlogo_norm(S,'alphabet','aa'); %# Use the modified SEQLOGO function
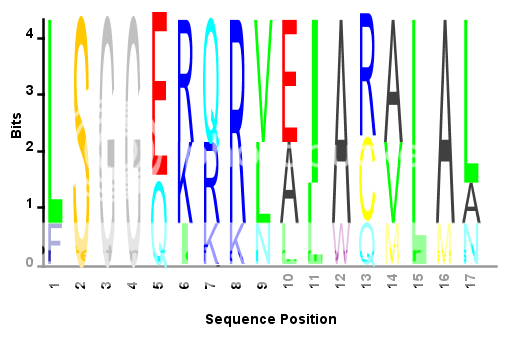 旧答案:
旧答案:
我不确定如何转换序列配置文件信息以从
Bioinformatics Toolbox函数SEQLOGO中获得所需的输出,但是我可以向您展示如何修改为
我对链接到
的相关问题的回答而编写的替代seqlogo_new.m 。如果您bitValues更改用于初始化bitValues的行:
bitValues = W{2}; 对此:
bitValues = bsxfun(@rdivide,W{2},sum(W{2})); 然后,您应该将每列缩放为1的高度。例如:
S = {'ATTATAGCAAACTA',... %# Sample sequence 'AACATGCCAAAGTA',... 'ATCATGCAAAAGGA'}; seqlogo_new(S); %# After applying the above modification
 更多&回答...
更多&回答...